
Type IX Glycogen Storage Disease
Synonyms:
Phosphorylase Kinase Deficiency
GSD type IX is a disorder in which the body cannot break down (metabolize) glycogen (a complex form of sugar). When someone has GSD IX, glycogen is stored in the organs of the body (liver, muscle and rarely heart) instead of being used. To briefly review metabolism, a simple form of sugar (glucose) is the bodies' main source of energy. After a person eats, there is too much glucose in the blood, so the body stores the extra glucose in the form of glycogen in the liver and muscles (much like storing extra food from the grocery store in the pantry to be used later, when needed). When the body needs more energy, specific proteins (enzymes) change the glycogen back to glucose and take it out of the liver and the muscles (just like taking food out of the pantry).
There are many different steps involved in breaking down glycogen into glucose. Many different enzymes help each step of breakdown happen in a sequential manner. Patients with type of GSD IX glycogen storage disease have a deficiency of the enzyme called phosphorylase kinase. Phosphorylase Kinase Deficiency, PHK, constitutes the largest subgroup (1:100,000 births) of liver glycogenosis. When phosphorylase kinase is low or deficient, glycogen cannot be broken down completely. The phosphorylase kinase enzyme is a regulatory enzyme in the breakdown of glycogen; thus, the deficiency of this enzyme results in glycogen accumulation. When there is a defect in phosphorylase kinase, the body may not be able to make enough sugar (glucose) to use, and also the incompletely digested glycogen builds up (accumulates) in the body, primarily in the liver and muscles. Although some glucose (energy) is created from the successful earlier steps in the breakdown of glycogen, not enough glucose is made for the proper function of the body.
The clinical picture of Type IX glycogen storage disease is similar to that seen in Type VI GSD, liver phosphorlyase deficiency. GSD IX can cause low levels of glucose in the blood (hypoglycemia or low blood sugar), which has the potential to lead to seizures. This is more likely to happen after long periods of not eating (fasting), and can be prevented by maintaining a high carbohydrate (starchy foods) diet, adequate amounts of protein in the diet, and avoiding long periods of not eating. A deficiency in PHK causes glycogen accumulation in various tissues and organs including liver, muscle, blood cells but rarely in the heart. The liver can become irritated or inflamed from storing increased amounts of glycogen, which can cause some blood tests to be abnormal with elevated enzymes (ALT and AST) on liver function blood tests. The most common symptoms are enlarged liver, growth retardation, mild delay in motor development, and elevated blood lipids (fats). The symptoms usually improve as a child ages, and children usually reach their full potential height and weight by adulthood.
Phosphorylase kinase enzyme is made up of four different pieces (subunits), which we can compare to puzzle pieces. When putting together a puzzle, the picture is not complete unless all of the pieces are placed together correctly. In the same way, all the pieces (subunits) of this enzyme must come together correctly in order for the enzyme to work properly. If a puzzle piece is missing or has the wrong shape, the puzzle cannot be completed. Likewise, if there is a change in one of the subunits, the enzyme cannot be assembled and formed properly and will not be able to perform its job in the breakdown of glycogen. The inheritance of Type IX glycogen storage disease can be either autosomal recessive or X-linked recessive.
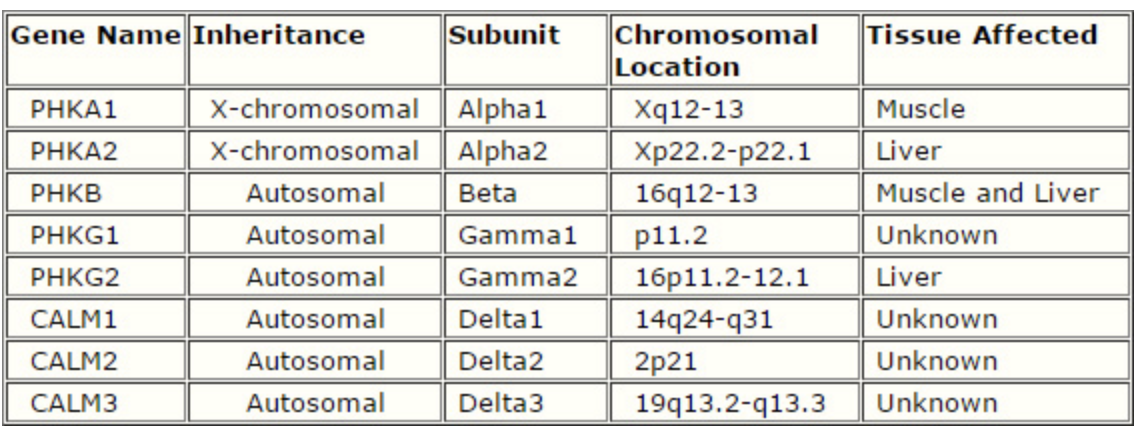
There are multiple genes that provide the instructions to make the four pieces/subunits (i.e. alpha, beta, gamma, and delta) that make up the phosphorylase kinase enzyme. A change in a subunit is caused by a change in the gene that gives the instructions for that subunit. A definitive diagnosis of PHK deficiency requires demonstration of the enzymatic loss in certain tissues (blood erythrocytes), muscle, heart and liver tissues). DNA testing is currently available for the subunits alpha1, alpha2, and gamma2. Not many patients have had genetic changes found in beta, delta1, and delta2.
The most common form of Type IX GSD is the X-linked form, which accounts for nearly 75% of all cases. X-linked recessive conditions mainly affect boys. The X-linked type involves genetic changes affecting the alpha-subunit of PHK genes (PHKA1 and PHKA2 genes). The main organs affected include the blood cells, muscle and liver. The clinical picture associated with these genetic changes is hepatomegaly (large liver), growth retardation, and mild to moderately elevated cholesterol and fat in blood and liver enzymes.
The most common autosomal recessive sub-types of GSD IX are caused by mutations in the PHKG2 gene or the PHKB gene. This form of GSD IX is inherited in an autosomal recessive manner. Although mutations in the PHKB gene can cause both liver and muscle glycogen accumulation, patients usually have liver complications. PHKB mutations tend to cause the mildest form of PHK deficiency. In contrast, individuals with mutations in the PHKG2 subunit of the PHK enzyme usually have more severe symptoms; PHKG2 gene changes are particularly associated with recurrent hypoglycemia, hepatomegaly and rarely liver fibrosis.
Currently, the only treatment for GSD IX is based on the symptoms of the condition. Hypoglycemia (low blood sugar) can be controlled by frequent meals high in carbohydrates with cornstarch supplements or night time stomach drip feedings if needed. As it is understood now, patients with mutations in the PHKA2 or PHKB genes typically have lessening of their symptoms by puberty. However, patients with more severe types and symptoms may need to remain on treatment regimens to avoid hypoglycemia.
As our understanding increases with GSD IX through long term research studies done on GSD IX patients, like other GSDs, the long term complications will be better understood. Research is still progressing and presenting more information about this disease. Prognosis (prediction of future health) is generally considered good for the liver forms of the disease; however, prognosis for the muscle forms is still unknown.
Summary of the genes involved in Phosphorylase Kinase Deficiency:
Additional Support:
Muscular Dystrophy Association [MDA] Resources: www.mdausa.org
Metabolic Diseases of Muscle
”Living with Metabolic Myopathies”
About the associationThe Association for Glycogen Storage Disease - AGSD - was established in 1979 in order to create an organization which would be a focus for parents of and individuals with glycogen storage disease (GSD) to communicate, share their successes and concerns, share useful findings, provide support, create an awareness of this condition for the public, and to stimulate research in the various forms of glycogen storage diseases. This website provides basic information about the glycogen storage diseases. The information is intended to be of use to people affected by one of the glycogen storage diseases, their families, and other interested parties. Some forms of GSD cause little in the way of illness, while others are life threatening. Included in this site is a description of the general symptoms, associated problems, current treatment, and long-term outcome for the most commonly diagnosed glycogen storage diseases. It does not do justice to the difficulty patients and their families' experience, and their deep desire for improved forms of treatment or ultimately total correction. | ContactAssociation for Glycogen Storage Disease 1542 Flammang Dr. PMB 1004 Waterloo Iowa 50702
|


